
Two New Mutations in Mitochondria DNA Associated with Isolated Myopathy
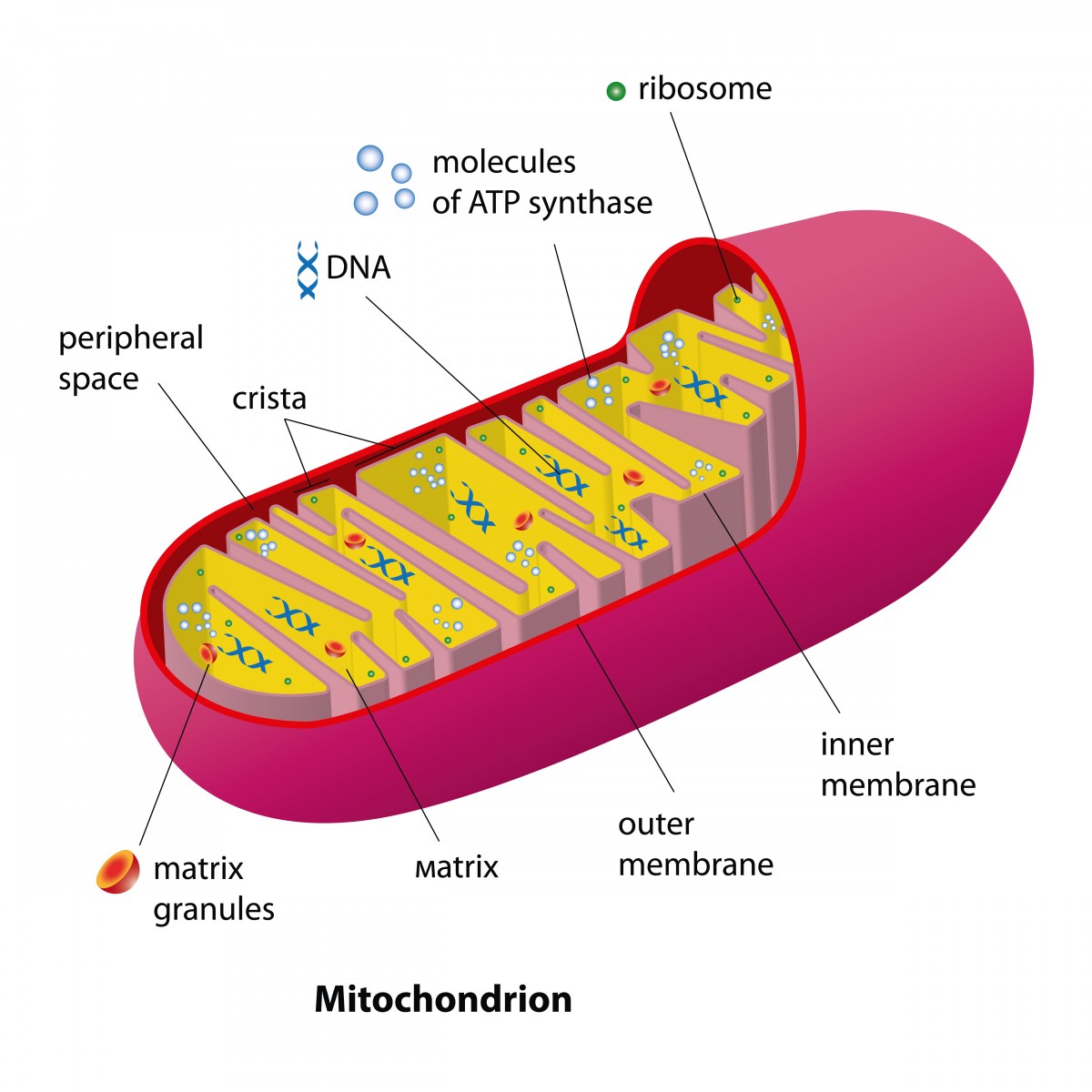



In a new study entitled “Pathogenic mitochondrial mt-tRNAAla variants are uniquely associated with isolated myopathy,” authors report to have identified in two patients with isolated myopathy (a muscular disease characterized by deficient muscle fibers resulting in muscular weakness) two novel variants in the mitochondria DNA, specifically in the mitochondria-tRNAAla gene, which are highly pathogenic and triggering the disease. The study was published in the European Journal of Human Genetics advanced online publication section of 15 April 2015.
Mitochondria, the organelle inside eukaryotic cells responsible for converting the energy within food into energy cells can use called ATP, also harbors a small portion of DNA, called mitochondrial DNA (mtDNA). Similar to the DNA packed inside a cells’ nucleus, mtDNA point mutations can lead to pathological phenotypes and mtDNA disorders display differences in disease severity, ranging from mild to severe. Previous studies reported that mutations in mt-tRNA genes, a class of transfer RNAs (tRNAs) coded by mt-DNA whose function is to help assemble protein building blocks (amino acids) into functioning proteins, are prevalent in adults and usually affect several organs. In this study, authors present two clinical cases where both patients carry novel variants in the mt-tRNAAla gene with pathogenic effects restricted to muscle fibbers resulting in myopathy.
[adrotate group=”4″]
Patients were both women who were 29 and 69 years old, respectively, presenting muscle weakness and with no history of muscle disease in their families. Authors performed standard histology analysis of patients’ muscle biopsies. Additionally, total DNA was extracted from muscle tissues and entire mitochondrial genome was analyzed by sequencing. The team found patients exhibited extensive muscle pathological alterations at the histological level, including muscle fibers deficient in cytochrome c oxidase (COX) activity, a marker of mitochondrial dysfunction, together with mitochondria accumulation. Sequencing analysis detected two novel mutations within the mt-tRNAAla gene that were confirmed as pathogenic, according to previously established criteria. The team also identified very high-mutation levels in all tissues (including muscle, urinary epithelia, buccal epithelia, hair shafts and blood) of each patient, but always the highest mutation load was observed in the muscle.
mt-tRNA mutations triggering isolated disease are rarely detected in the clinics. However, the mutations here identified in the mt-tRNAAla gene were all associated with isolated myopathy, while previous mutations in other mt-tRNA genes are usually associated with multi-organ disease.


