
Mitochondrial Replacement Therapy Shows Promise at Preventing Disease, But Challenges Remain



Because treatment for mitochondrial DNA diseases is largely limited to addressing only symptoms, researchers are increasingly looking at mitochondrial replacement therapies as a way to allow women to have genetically related children while reducing the risk of transmitting disease.
Mitochondrial replacement therapies involve a new and innovative technology to prevent the inheritance of mutations in mitochondrial DNA. But the technology still faces challenges before it becomes widely available, according to a recent column published in the journal Nature Reviews Molecular Cell Biology. The column is titled, “Progress in mitochondrial replacement therapies.”
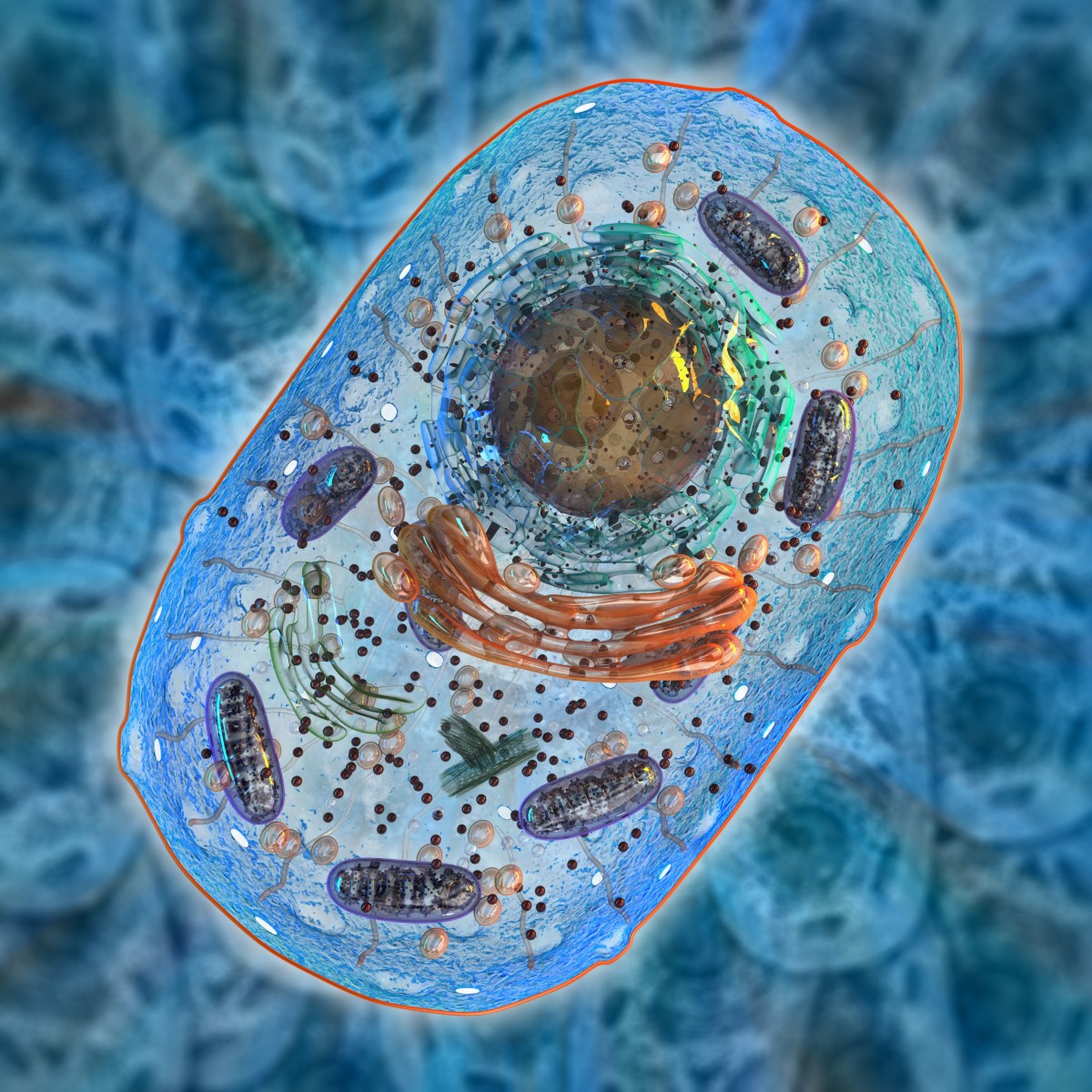
People have two types of DNA in their cells. Nuclear DNA, which represents the vast majority of a cell’s DNA, is located in the nucleus. Additionally, a small amount of DNA is present in the mitochondria, called mitochondrial DNA (mtDNA).
Unlike nuclear DNA, which is inherited from both parents, mtDNA is inherited from the mother only. Mothers always pass mtDNA mutations, which are genetic errors or defects, to their children, while fathers never pass them on. mtDNA diseases are fairly common, affecting about 1 in 5,000 people.
Because multiple copies of mtDNA exist in all cells, many patients with these diseases tend to have a mixture of mutated and normal mtDNA, a phenomenon known as heteroplasmy. It becomes challenging, therefore, to provide reproductive advice to families with heteroplasmic DNA mutations, as children often have considerable variation in mtDNA mutations.
While for some couples, prenatal or pre-implantation genetic diagnosis to select embryos with low levels of mtDNA mutation is an option, for women with high levels of heteroplasmy or homoplasmy, in which all mtDNA is mutated, options are limited.
Mitochondrial replacement therapy involves transplanting the nuclear DNA from the egg of an affected woman to an enucleated egg — an egg without a nucleus — from a healthy donor. This can be performed either before or after fertilization.
Two potential methods of conducting mitochondrial replacement therapy exist, called pronuclear and metaphase II spindle transfer, referring to the different developmental stages at which the procedure takes place.
Proof-of-concept studies for pronuclear and metaphase II spindle transfer indicate that both procedures can lead to the development of early-stage human embryos with minimal transfer of maternal mtDNA. Levels of mtDNA carryover ranged from 0-5% with the pronuclear method and 0-2% in the spindle transfer method.
Despite the low levels of mtDNA carryover, some cells appeared to revert to the “minority” genome, which in this case is the mother’s mutated genome. It is unclear how significant this would be clinically, and scientists say it is important to further minimize the contribution of maternal mtDNA.
While significant research suggests that mitochondrial replacement therapy is ready for clinical evaluation, risks associated with this approach still exist. One of them that scientists are discussing is the potential that nuclear and mitochondrial DNA could be a haplogroup mismatch, similar to when patients undergoing organ transplant reject the organ. However, scientists believe this risk likely is minimal.
Another challenge is that supplies of human eggs for research are limited.
The therapy also faces ethical and legislative challenges due to concerns that this technique could lead to a slippery slope of allowing DNA alterations.
In the U.K., after extensive ethical reviews, public consultations, four separate independent scientific reviews, and debates in both houses of Parliament, the therapy was given the green light in 2016. The first license for a center to proceed with mitochondrial replacement therapy was approved last year.


