
Mutation in YME1L1 Gene Linked to Mitochondrial Disease for 1st Time



Researchers at the Charité University Medicine Berlin in Germany have, for the first time, shown that a mutation in the YME1L1 gene is linked to mitochondrial disease, identified in four children born to related parents.
The study, “Homozygous YME1L1 mutation causes mitochondriopathy with optic atrophy and mitochondrial network fragmentation,” published in the online journal eLife, shows that the protein YME1L1 is crucial for the proper function of mitochondria. More studies are needed for a fuller understanding of the spectrum of symptoms the mutation can give rise to.
Four children, born without any complications and seemingly healthy at birth, were soon obviously found not to be following a normal developmental path. All had mental disabilities, movement problems, delays in speech development, and difficulties with both hearing and eyesight linked to a breakdown of the optic nerve.
They had normal facial features, but some had abnormally small heads, little control of bodily movements or muscle spasms, and writhing or stereotypic movements. One child had an abnormally large head.
Brain scans revealed that each of the four had lesions in the white matter of the brain, and two of them had signs of tissue loss. Three had higher levels of lactate and higher lactate/pyruvate ratios than normal, a typical sign of mitochondrial disease.
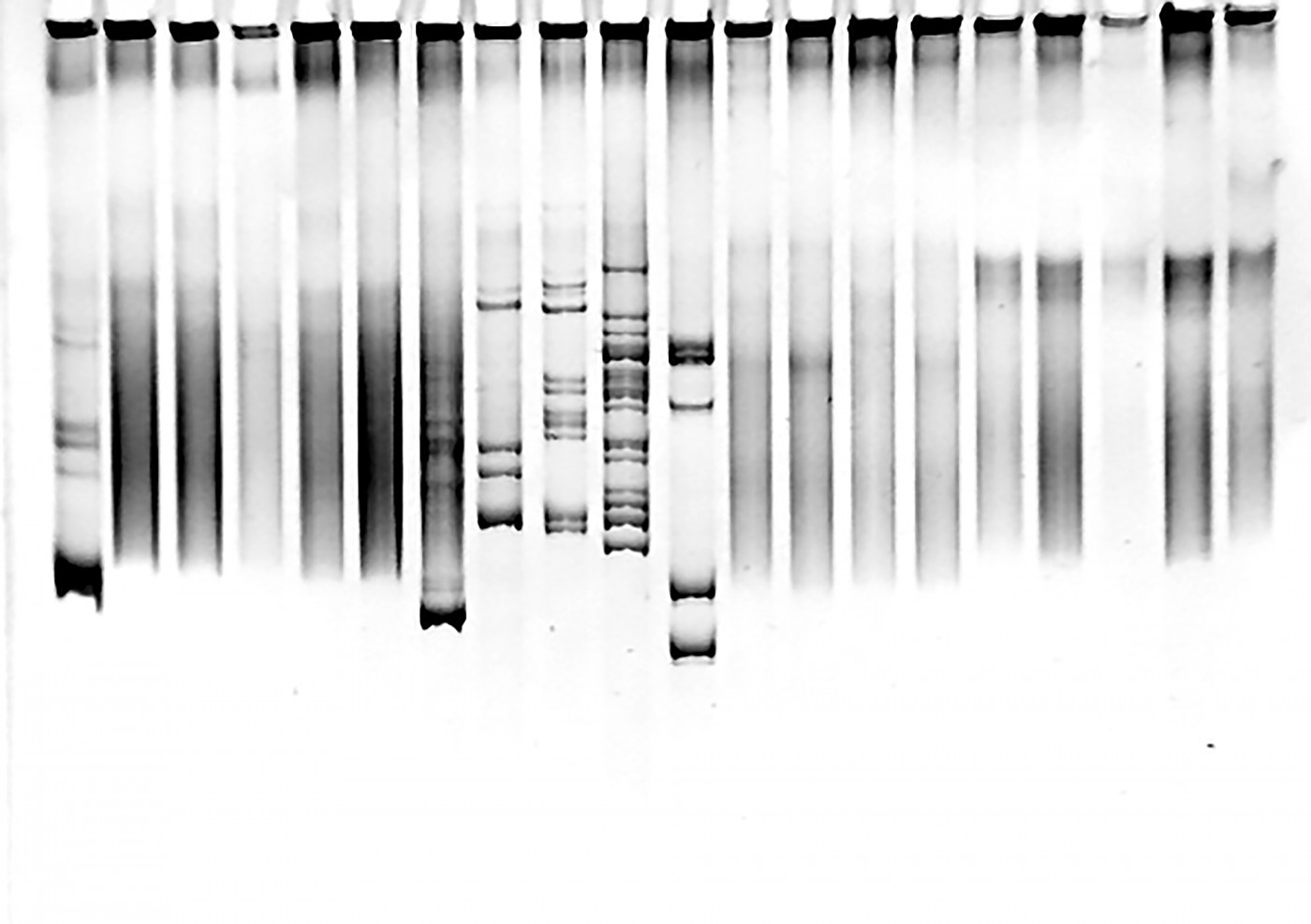
A muscle biopsy from one of the children revealed abnormal-looking mitochondria, so physicians tested the four children for a range of known genetic conditions, including sequencing the mitochondrial DNA.
These initial screens did not reveal any abnormalities. Researchers then embarked on sequencing all the coding parts of the genome, so-called whole exome sequencing. This analysis revealed that all children had a mutation in both gene copies of the YME1L1 gene.
In trying to understand how this mutation contributes to disease, the team performed extensive molecular testing. The protein formed by the gene is inserted into the inner mitochondrial membrane. First, however, an early version of the protein needs to be cleaved. Researchers found that the mutation prevented the cleavage from occurring. Thus, instead of being incorporated into mitochondria, the protein was recognized as a misfolded protein by the cellular machinery and destroyed. This, in turn, prevented the proper function of the protein OPA1, leading to fragmentation of the mitochondrial networks.
“Impaired YME1L1 function causes a proliferation defect and mitochondrial network fragmentation due to abnormal processing of OPA1. Our results identify mutations in YME1L1 as a cause of a mitochondriopathy with optic nerve atrophy, highlighting the importance of YME1L1 for mitochondrial functionality in humans,” the research team concluded.


